Input data and parameters
QualiMap command line
| qualimap bamqc -bam E2348-69.bam -c -nw 400 -hm 3 |
Alignment
| Command line: | "/usr/local/ngseq/packages/Aligner/Bowtie2/2.5.4/bin/bowtie2-align-s --wrapper basic-0 -p 8 --rg-id E2348-69 --rg SM:E2348-69 --rg LB:RGLB_E2348-69 --rg PL:illumina --rg PU:RGPU_E2348-69 -x /srv/GT/reference/Escherichia_coli_O127H6_EPEC-E2348_69-V2/Ensembl/EPEC-E2348_69-V2/Sequence/BOWTIE2Index/genome -1 /scratch/Bowtie2_2025-02-06--09-29-00_E2348-69_temp3385756/E2348-69-trimmed_R1.fastq.gz -2 /scratch/Bowtie2_2025-02-06--09-29-00_E2348-69_temp3385756/E2348-69-trimmed_R2.fastq.gz" |
| Draw chromosome limits: | yes |
| Analyze overlapping paired-end reads: | no |
| Program: | bowtie2 (2.5.4) |
| Analysis date: | Thu Feb 06 09:42:59 CET 2025 |
| Size of a homopolymer: | 3 |
| Skip duplicate alignments: | no |
| Number of windows: | 400 |
| BAM file: | E2348-69.bam |
Summary
Globals
| Reference size | 4,848,621 |
| Number of reads | 5,827,464 |
| Mapped reads | 5,536,810 / 95.01% |
| Unmapped reads | 290,654 / 4.99% |
| Mapped paired reads | 5,536,810 / 95.01% |
| Mapped reads, first in pair | 2,772,492 / 47.58% |
| Mapped reads, second in pair | 2,764,318 / 47.44% |
| Mapped reads, both in pair | 5,516,332 / 94.66% |
| Mapped reads, singletons | 20,478 / 0.35% |
| Secondary alignments | 0 |
| Read min/max/mean length | 18 / 151 / 135.12 |
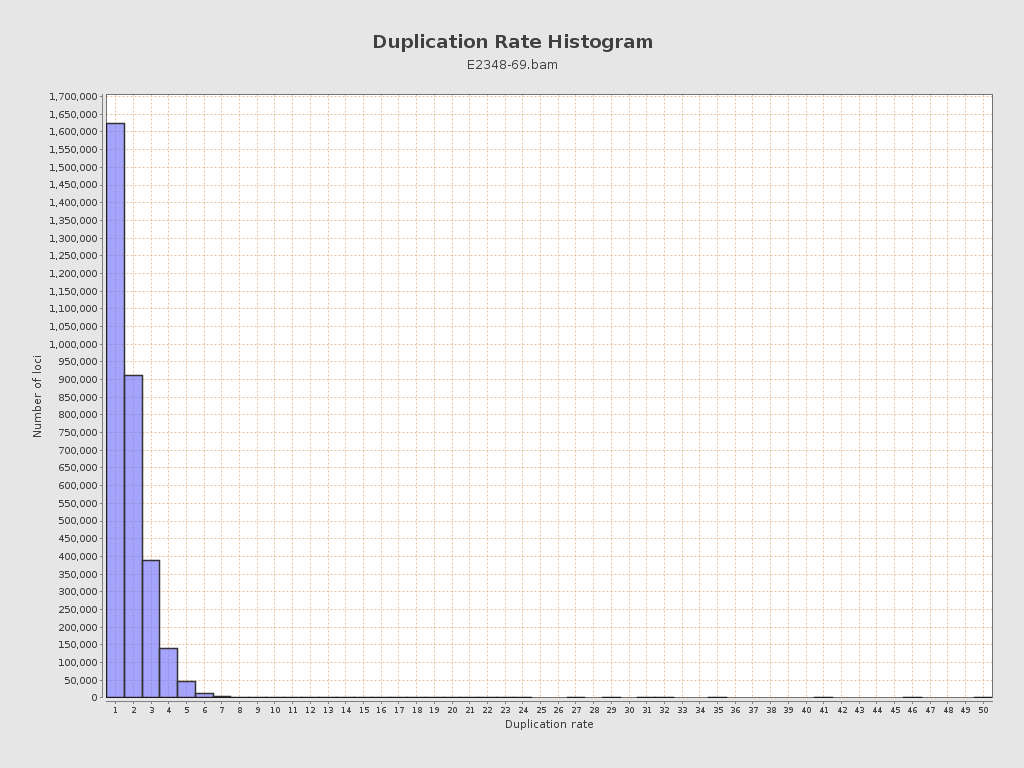
| Duplicated reads (flagged) | 184,143 / 3.16% |
| Clipped reads | 0 / 0% |
ACGT Content
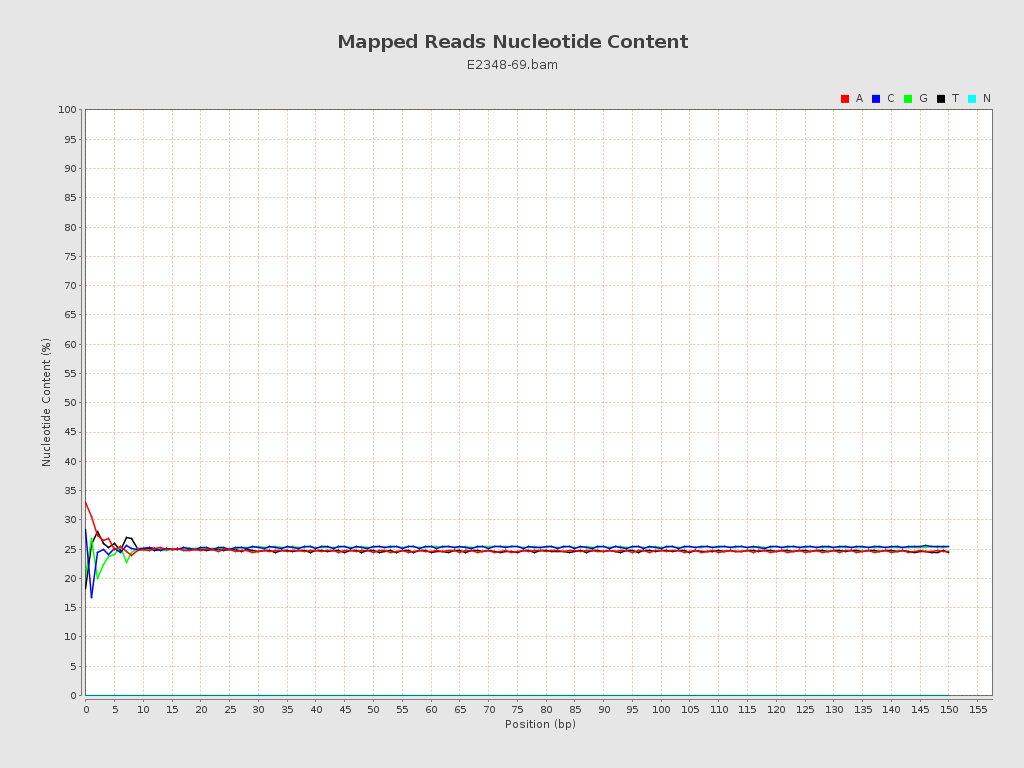
| Number/percentage of A's | 186,015,354 / 24.85% |
| Number/percentage of C's | 189,022,682 / 25.25% |
| Number/percentage of T's | 185,151,655 / 24.73% |
| Number/percentage of G's | 188,459,333 / 25.17% |
| Number/percentage of N's | 14,173 / 0% |
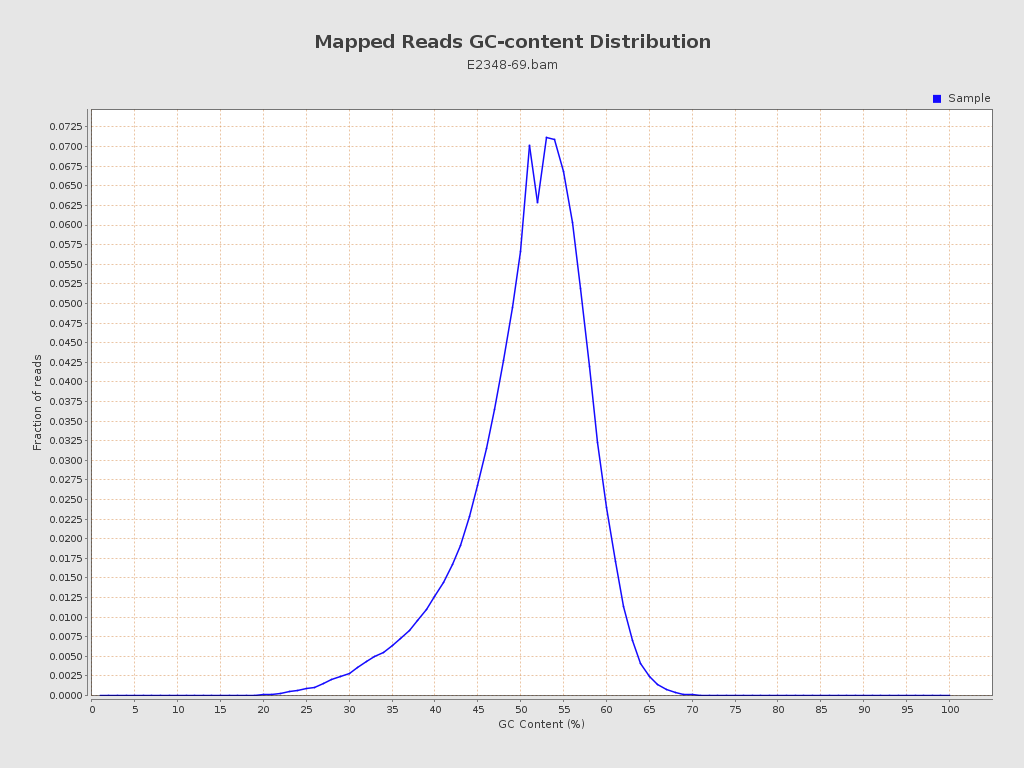
| GC Percentage | 50.42% |
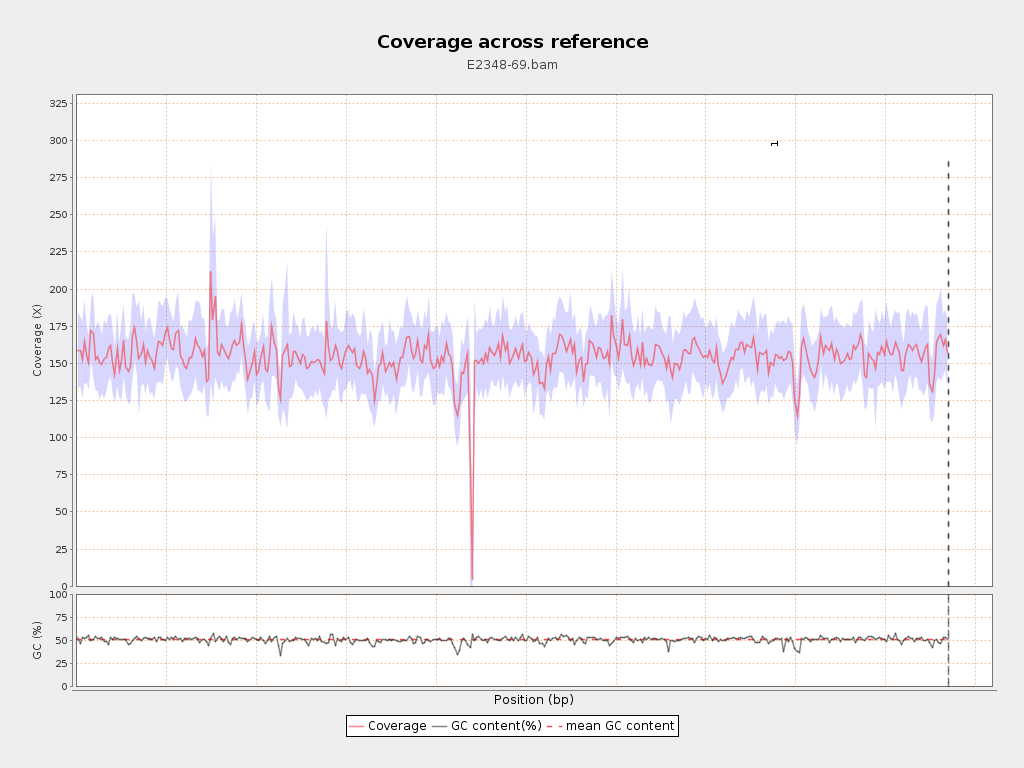
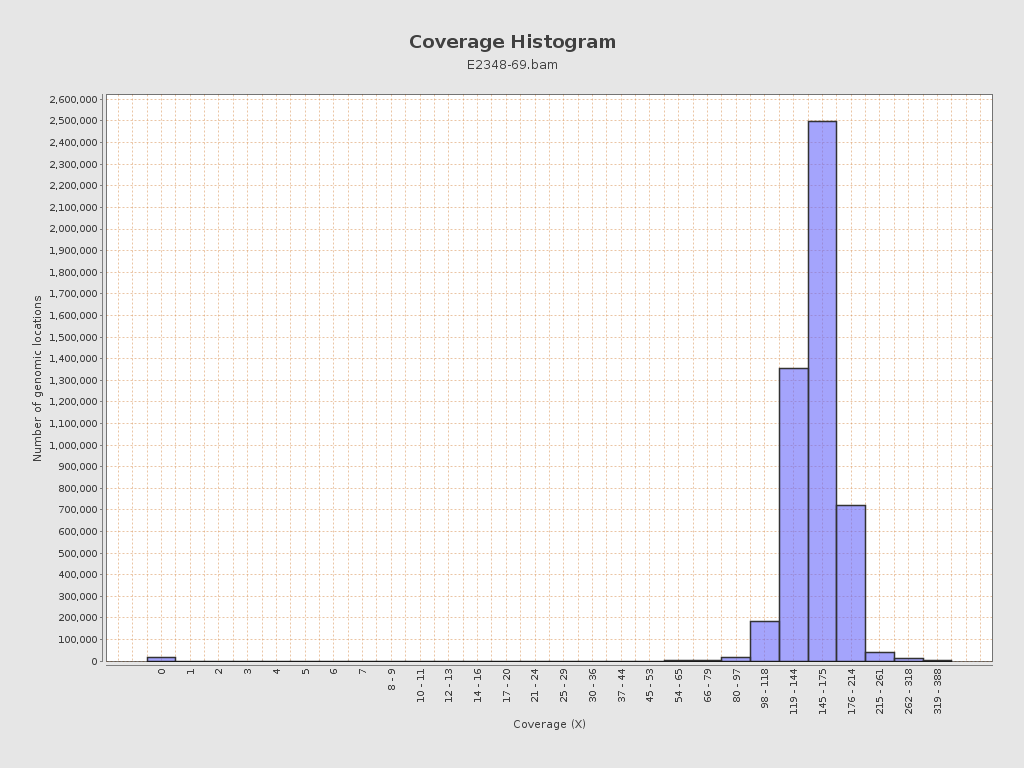
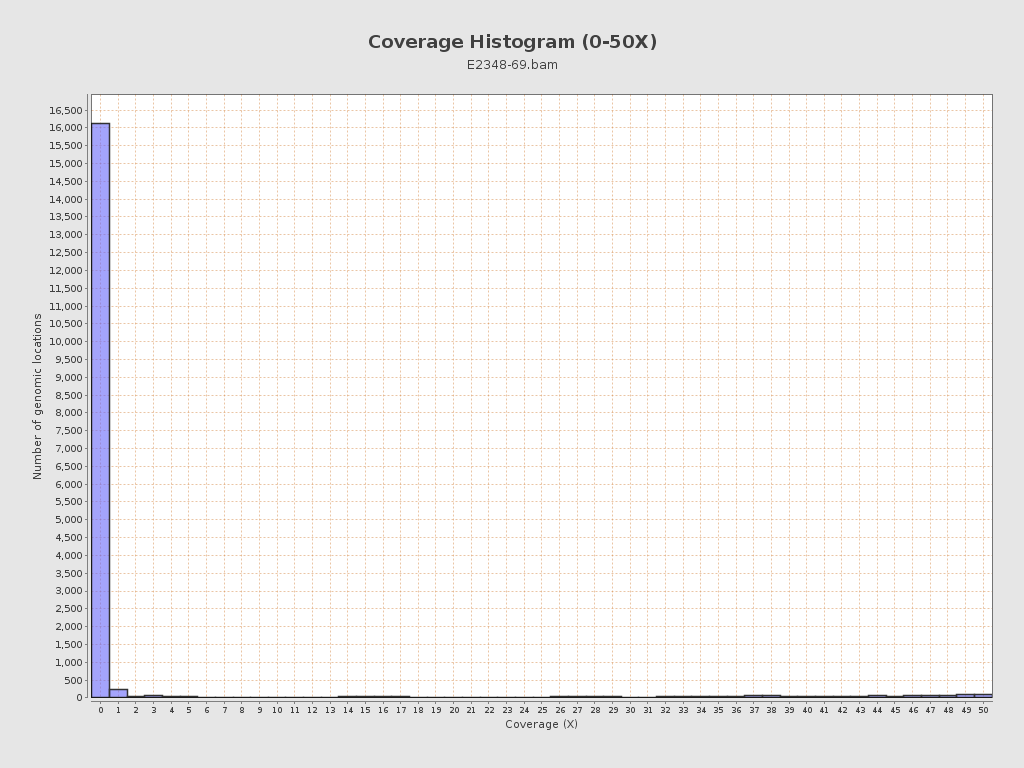
Coverage
| Mean | 154.4098 |
| Standard Deviation | 25.1437 |
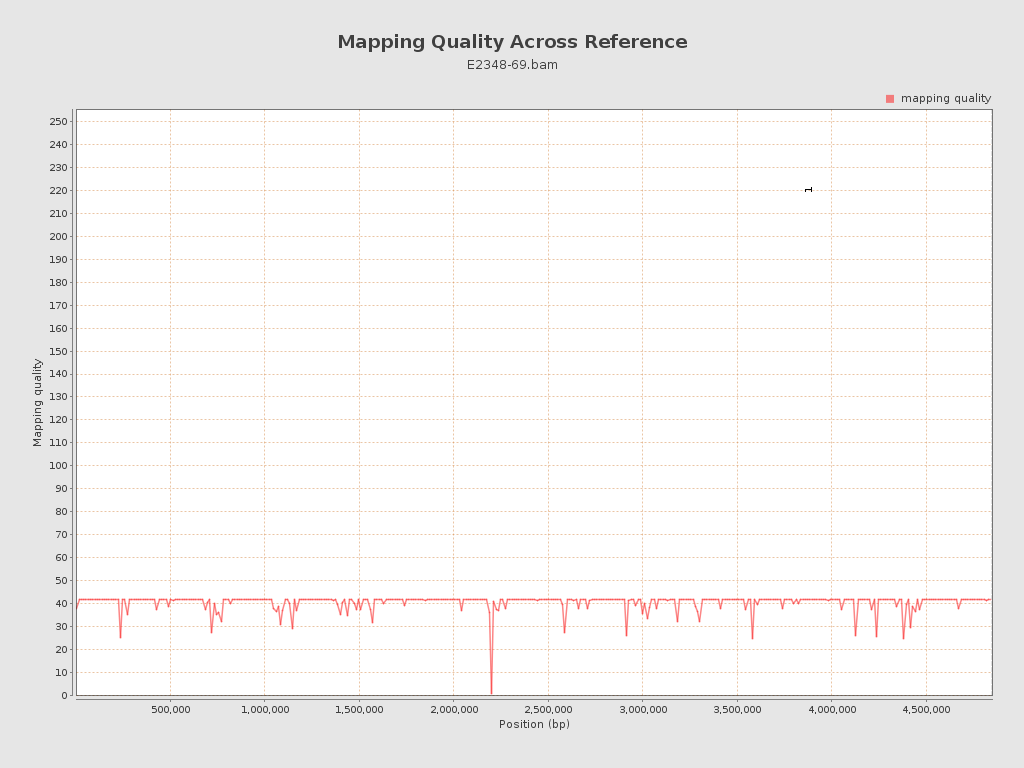
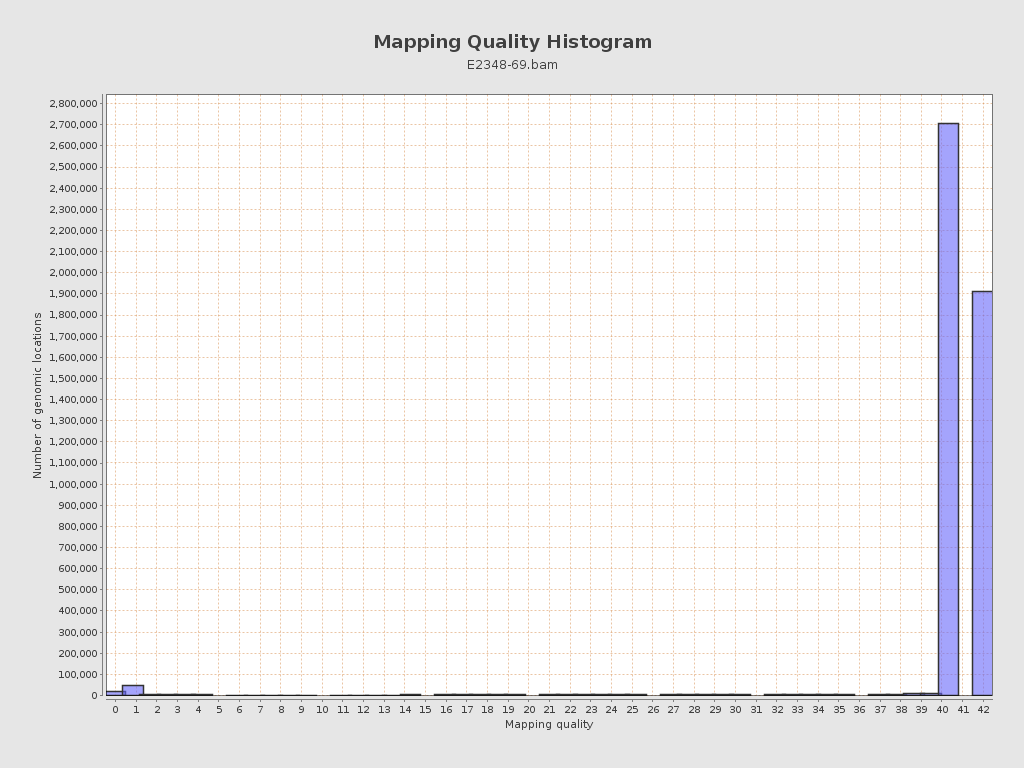
Mapping Quality
| Mean Mapping Quality | 40.75 |
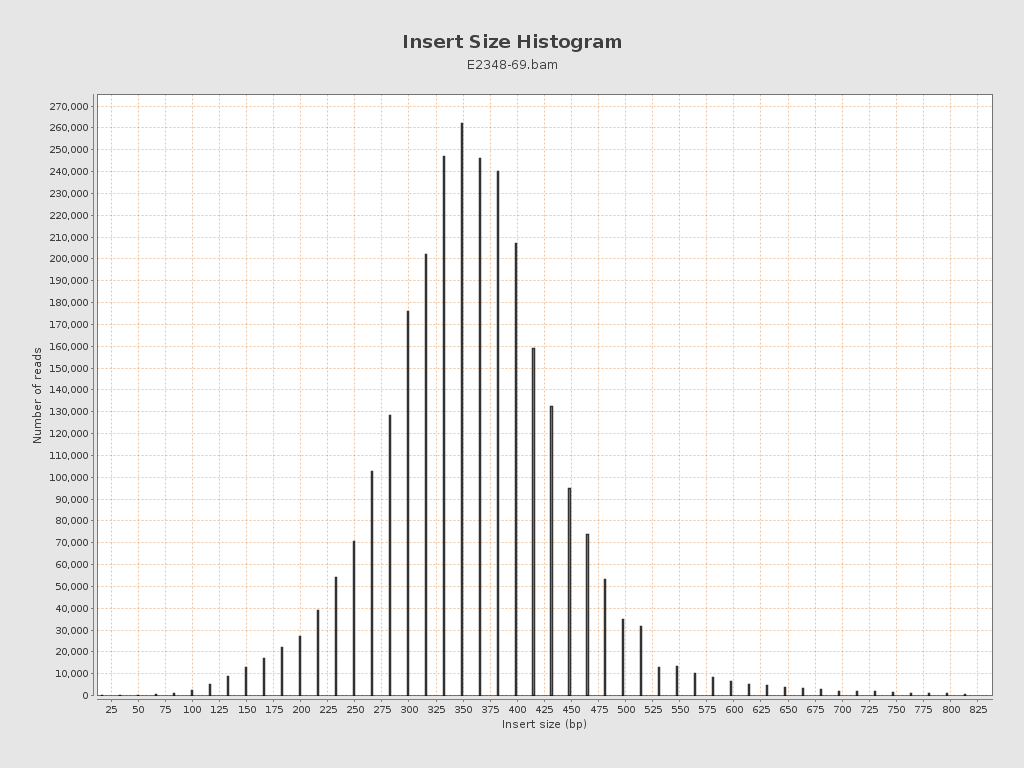
Insert size
| Mean | 9,798.86 |
| Standard Deviation | 152,850.96 |
| P25/Median/P75 | 317 / 365 / 415 |
Mismatches and indels
| General error rate | 0.19% |
| Mismatches | 1,265,643 |
| Insertions | 29,263 |
| Mapped reads with at least one insertion | 0.4% |
| Deletions | 9,155 |
| Mapped reads with at least one deletion | 0.16% |
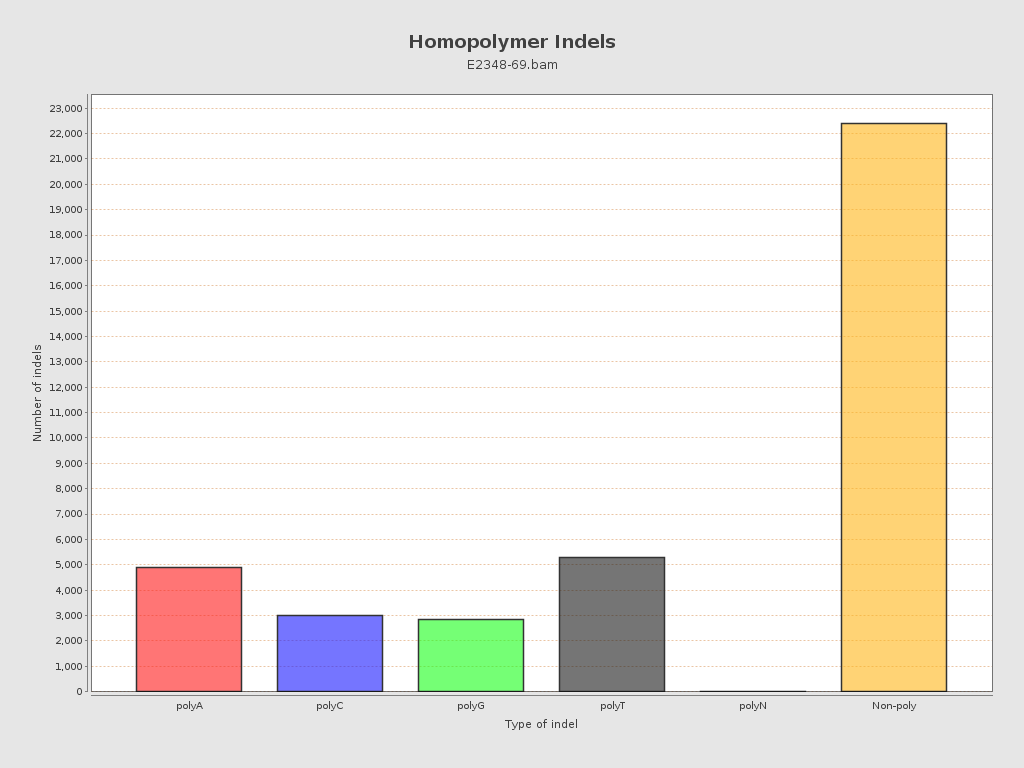
| Homopolymer indels | 41.68% |
Chromosome stats
| Name | Length | Mapped bases | Mean coverage | Standard deviation |
| 1 | 4848621 | 748674691 | 154.4098 | 25.1437 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}